deepEA User Manual
(version 1.0)- deepEA is a convenient, freely available, web-based platform that is capable to support deep analysis of epitranscriptome sequencing data with several general and specific functionalities. deepEA consists of six modules: Data Preparation, Quality Control, Identification of RNA Modifications, Functional Annotation, Multi-omics Integrative Analysis and Prediction Analysis Based on Machine Learning.
- deepEA project is hosted on https://cma2015.github.io/deepEA.
- deepEA docker image is available in https://hub.docker.com/r/malab/deepea.
- The deepEA demo server can be accessed via http://deepea.omicstudio.cloud.
- The following part shows installation of deepEA docker image and detailed documentation for each function in deepEA.
Identification of RNA Modifications
This module provides step-by-step functions required for epitranscriptome reads mapping and identification of RNA modifications.
Align Reads to Genome
Several commonly used aligners are wrapped to align epitranscriptome reads to genome. Currently, Tophat2, Bowtie2, STAR, HISAT2, bwa-mem.
| Tools | Description | Input | Output | Time (test data) | Reference |
| Tophat2 | Tophat2 is a spliced aligner, which aligns short reads by calling Bowtie2 but alows for variable-length indels with respect to the reference genome. | Epitranscriptome sequencing reads in FASTQ format and reference genome sequences in FASTA format | Read alignments in SAM/BAM format | ~50s | Kim et al., 2013, Genome Biology |
| Bowtie2 | Bowtie2 is a short read aligner which achieves a combination of high speed, sensitivity and accuracy by combining the strengths of the full-text minute index with the flexibility and speed of hardware-accelerated dynamic programming algorithms, therefore bowtie2 is suitable for large genomes | ~10 s | Langmead et al., 2012, Nature Methods | ||
| STAR | STAR is an ultrafast universal RNA-Seq aligner and can discover non-canonical splices and chimeric (fusion) transcripts | ~16s | Dobin et al., 2013, Bioinformatics | ||
| HISAT2 | HISAT2 is an ultrafast spliced aligner with low memory requirements. It supports genomes of any size, including those larger than 4 billion bases | ~8s | Kim et al., 2015, Nature Methods | ||
| bwa-mem | bwa-mem is a relatively early aligner based on backward search with Burrows–Wheeler Transform | ~10s | Li et al., 2009, Bioinformatics |
Identify RNA Modifications
Identify RNA Modifications implements three pipelines for MeRIP-Seq, CeU-Seq and RNA-BSSeq, respectively.
| Tools | Description | Input | Output | Time (test data) | Reference |
|---|---|---|---|---|---|
| Peak Calling from the MeRIP-Seq data | Identify enriched genomic regions from MeRIP-Seq experiment | Read alignments of IP and input in SAM/BAM format and reference genome sequences in FASTA format | RNA modifications in BED format | ~36s | Zhai et al., 2018, Bioinformatics |
| Calling m5C from the RNA-BSseq data | Perform bisulfite sequencing (BS-Seq) read mapping, comprehensive methylation calling using meRanTK | Sequencing reads in FASTQ format and reference genome sequences in FASTA format | m5C sites in BED format | ~10 mins using 2 threads | Rieder et al., 2016, Bioinformatics |
| Calling Ψ from CeU-Seq data | Identify pseudouridylation from CeU-Seq | Read alignments in SAM/BAM format and cDNA sequences in FASTA format | Pseudoridylation sites in BED format | ~1 mins | Li et al., 2015, Nature Chemical Biology |
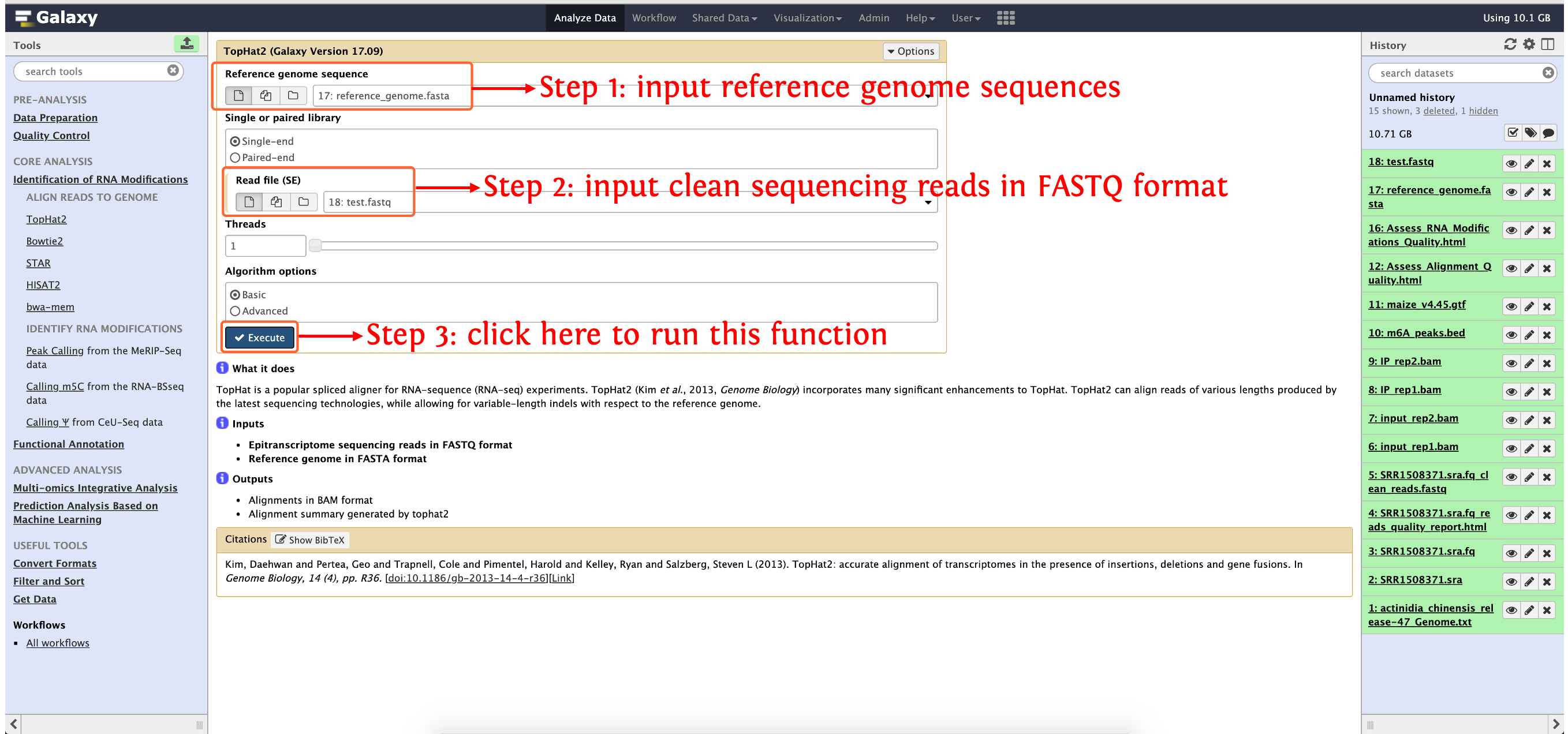
Align reads to genome
Currently, deepEA wrapped five aligners to map epitranscriptome reads to genome, here, we take Tophat2 as an example to show how to use deepEA to run reads mapping, the other four aligners are similar.
Input
- Epitranscriptome sequencing reads in FASTQ format
- Reference genome in FASTA format
Output
- Alignments in BAM format
- Alignment summary generated by tophat2
How to use this function
Step 1: upload the data in directory
test_data/Identification_of_RNA_Modifications/Align_Reads_to_Genome/to history panel, if you are not clear about how to upload local data to deepEA server, please see here for detailsStep 2: see the following screenshot to run this function
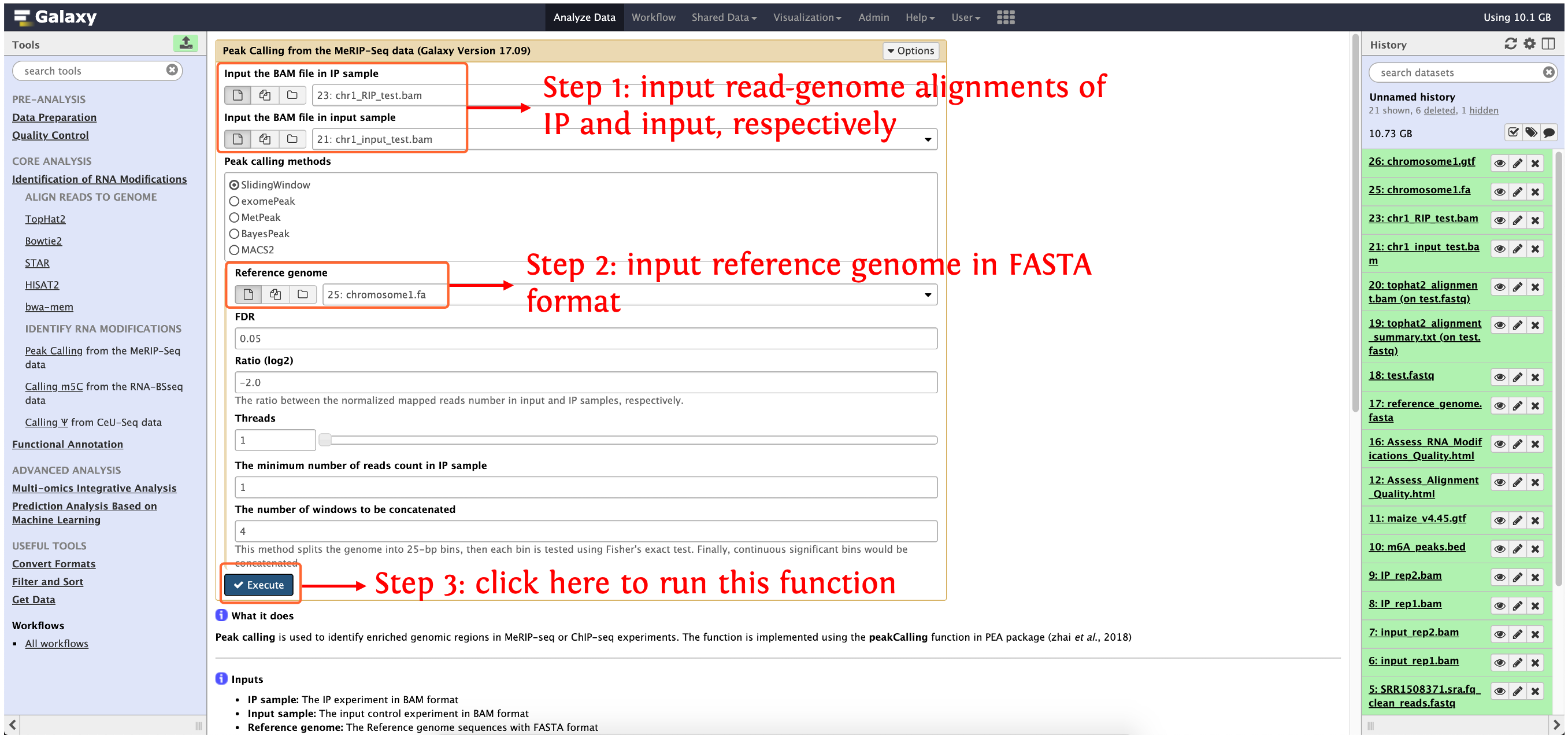
Peak calling from the MeRIP-Seq data
Peak calling is used to identify enriched genomic regions in MeRIP-seq or ChIP-seq experiments. The function is implemented using the peakCalling function in PEA package (zhai et al., 2018)
Input
- IP sample: The IP experiment in BAM format
- Input sample: The input control experiment in BAM format
- Reference genome: The Reference genome sequences with FASTA format
- Reference annotation file: The Reference genome annotation file with GTF/GFF3 format (required for methods: exomePeak, MeTPeak and BayesPeak)
Output
The enriched peak region matrix in BED format
For SlidingWindow method:
Chromosome Start(1-based) End Bin number Mean FDR Max FDR Minimum FDR Mean Ratio Max Ratio Minimum Ratio 1 67476 67575 4 0.0136 0.0328 0.0001 -1.0012 -0.6334 -1.581 1 330776 330875 4 0.0215 0.0381 0.0007 -1.576 -1.4077 -1.788 1 389201 389300 4 0.0024 0.0070 0.0002 -1.115 -1.0598 -1.190 For exomePeak metod:
Chromosome Start (0-based) End Gene ID P.value Strand 1 30663 30723 AT1G01040 0.0026 + 1 73831 74096 AT1G01160 2.5e-30 + 1 117530 117710 AT1G01300 2.4e-07 + For MetPeak method: it's the same as exomePeak
For BayesPeak method:
chr start end PP job 1 3748 3848 0.0231 2 1 6848 6948 0.0178 2 1 6898 6998 0.9960 1 For macs2 method: please see macs2
How to use this function
- Step 1: upload the data in directory
test_data/Identification_of_RNA_Modifications/Peak Calling from the MeRIP-Seq data/to history panel, if you are not clear about how to upload local data to deepEA server, please see here for details - Step 2: see the following screenshot to run this function
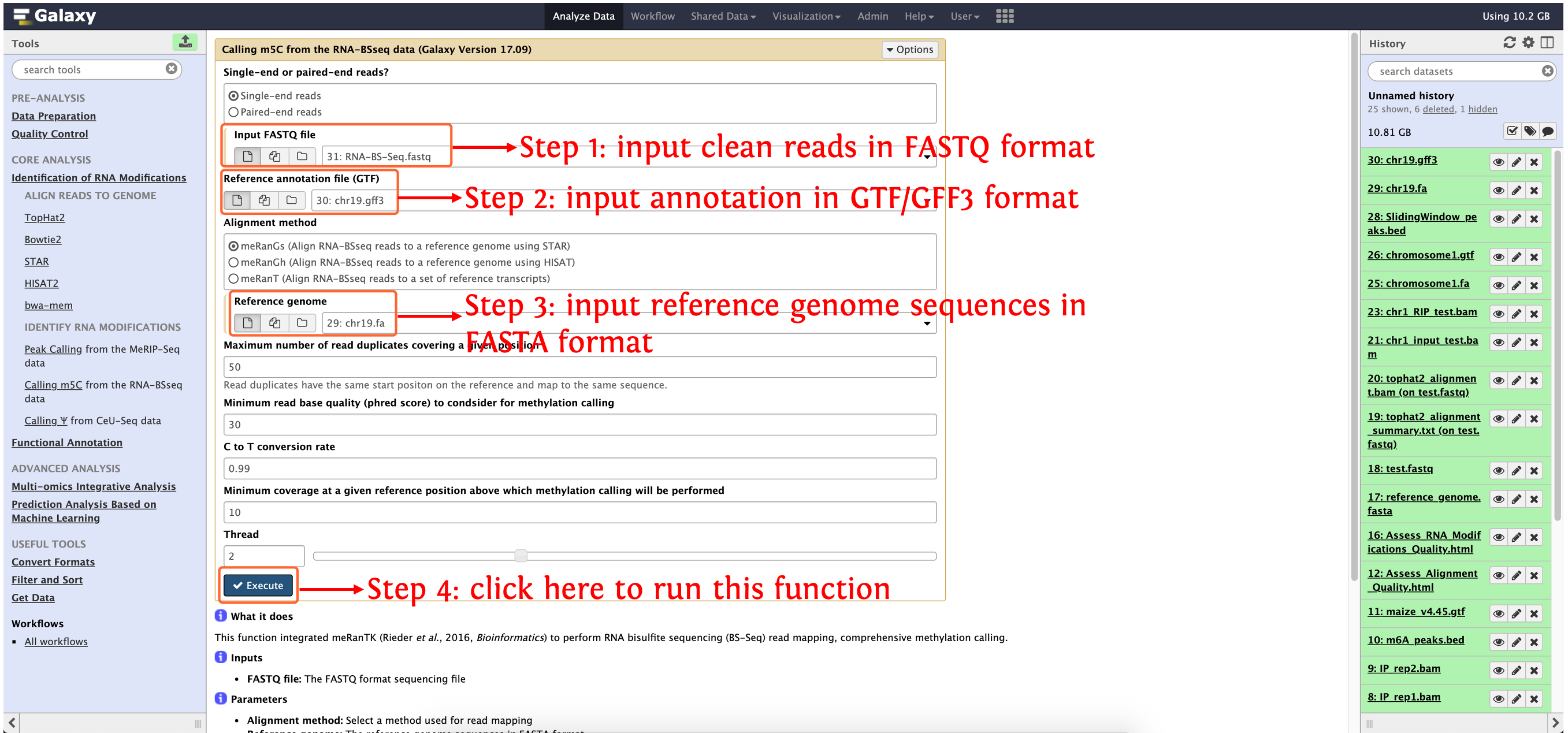
Calling m5C from the RNA-BSseq data
This function integrated meRanTK (Rieder et al., 2016, Bioinformatics) to perform RNA bisulfite sequencing (BS-Seq) read mapping, comprehensive methylation calling.
Input
- FASTQ file: The FASTQ format sequencing file
Output
- m5C_out_peaks: The detected m5C sites
How to use this function
- Step 1: upload the data in directory
test_data/Identification_of_RNA_Modifications/Calling m5C from the RNA-BSseq data/to history panel, if you are not clear about how to upload local data to deepEA server, please see here for details - Step 2: see the following screenshot to run this function
Calling Ψ from CeU-Seq data
This function is used to identify pseudouridylation from CeU-Seq (Li et al., 2015). To be specific, for any given position on a reference transcript, the stop rate of position i was calculated using the equation N_i_stop/(N_i_stop + N_i_readthrough), where N_i_stop (stop reads) is the number of reads with the mapping position starting at base i+1 (one nucleotide 3′ to position i), and N_i_readthrough (readthrough reads) is the number of reads reading through position i; Then a position i is identified to be Ψ only when all of the following criteria were met:
- the stop reads of position i (N_i_stop) must be no less than 5 in the N3-CMC(+) sample;
- the stop rate in N3-CMC(−) samples must be less than 0.10;
- the difference of stop rate for position i between the N3-CMC(+) samples and the matched N3-CMC(−) samples must be at least 0.30.
Input
- Pulldown sample in BAM format: The pulldown sample in BAM format
- Input sample in BAM format: The input sample in BAM format
- Input transcriptome in FASTA format: The transcriptome in FASTA format
Output
- A matrix containing the candidate pseudouridine sites
How to use this function
- Step 1: upload the data in directory
test_data/Identification_of_RNA_Modifications/Calling pseudouridylation from CeU-Seq/to history panel, if you are not clear about how to upload local data to deepEA server, please see here for details - Step 2: see the following screenshot to run this function
